UNEC Journal of Engineering and Applied Sciences Volume 2, No 2, pages 64-72 (2022) Cite this article, ![]() 1654
1654
Extraction, separation and raw material production using known and new extraction technologies from plants with target-oriented and multi-purpose production models is the most important step for domestic production in the field of food supplements and drug productions. In this context, an economical extraction model with multi-functional and target-oriented processing of plants reveals the most important and original value of this presentation. In this context, research and production studies for the extraction will be mentioned and the results of the studies carried out within the framework of the production model will be mentioned with a different perspective.
Plant-derived extracts and isolated natural compounds are valuable plant-derived products that have the potential to be used in food, medicine, cosmetics and different health fields, containing hundreds of powerful antioxidant, antimicrobial, antifungal and bioactive chemical components. However, new and innovative studies should be carried out to develop new extraction methods, industrial scale extraction and production models in order to obtain functional products with activity-oriented fractionation and to transform them into products with high added value.
Propolis is the general name of the resinous substances collected by honey bees from various natural plant sources, whose color can vary from dark brown to green and yellow depending on the origin and age of the bee [1]. In different studies, at least 200 different compounds such as phenolic and fatty acids and their esters, stilbene derivatives, substituted phenolic esters, terpenes, naphthalene, flavonoids, aromatic compounds, sesquiterpenes and steroids were determined from propolis samples [2].
Propolis is widely used among the public because it shows antioxidant, hepatoprotective, antimicrobial, antiprotozoal, antitumor, antibacterial, antifungal, antiviral, anti-inflammatory, anticancer and immunostimulant activities [3]. In addition, it has been determined that propolis inhibits hyaluronidase activity by delaying aging in cells [4, 5].
In this study, a new natural method was used by extracting propolis with water at different and low temperatures (+4°C, -80°C and -196°C). Freezing, pulverizing and extracting propolis at low temperatures without changing its structural integrity has not been applied in the scientific literature [6]. The aim of this study is to contribute to scientific knowledge of the phenolic, antioxidant, enzyme inhibitory, antibacterial and DNA protective properties of water-based Turkish propolis (WBTP) extracts.
Extraction of propolis
Natural propolis samples from the Western Black Sea Region of Turkey (Samsun, E:41.5051700 and W:36.0512400) in the natural environment of bees and hives were collected by professionals using sterile techniques. 100 g propolis samples were stored for 30 days at different conditions such as +4 °C (refrigerator), -80 °C (deep freezer) and -196 °C (liquid nitrogen). Propolis samples were made by thoroughly pulverizing using a laboratory grinder. For WBTP extracts, 50 g of PP was added to 500 mL of distilled distilled water at +4 °C (PP+4), -80 °C (PP-80) and -196 °C (PP-196). 500 mL of water was mixed in a glass bottle using a magnetic stirrer, then a homogenizer (Silverson, STL2) was added and mixing was continued. The homogeneous mixtures were filtered through a steel sieve. WBTP extracts were lyophilized at -50 °C under 0.01 mBr pressure to remove water (Christ Lyophilizer, Alpha 1-2 LD). Crude dried WBTP extracts were placed in a sterile medium and stored at -20 °C. [6]
Total phenol and total flavonoid content determination
The total phenol content (TPC) of WBTP extracts was determined using the Folin-Ciocalteu method [7]. We added 0.1 mL of sample extracts to sterile water containing FolinCiocalteu reagent and Na2CO3. The solutions were placed in tubes and incubated for 30 minutes in the dark. The absorbance of the reaction mixture was measured at 750 nm. The TPC of the WBTP extract was calculated from the gallic acid (GA) standard calibration curve (y=5.0012x+0.1505, R2 :0.99) to compare the absorbance values of the samples. mg gallic acid equivalent/g extract (mg GAE/g extract) was measured as dry weight. [6]
Total flavonoid content (TFC) was analyzed by the aluminum chloride method with modifications[7]. The extracts were mixed with NaNO2 and AlCl3 solutions in test tubes. After 5 minutes, NaOH was added to the reaction and incubated at 25 °C. The absorbance of the mixture was measured at 415 nm. TFC was calculated from the quercetin (QE) standard calibration curve (y=10.201x+0.0148, R2 : 0.99) to evaluate the absorbance values of the samples. Quercetin equivalents (QE) per mg dry weight were expressed as extract (mg QE/g extract) using standard quercetin.[6]
HPLC–MS/MS analysis
The chemical composition of WBTP extracts is equipped with Reverse-phase HPLC, a dual pump (1260 Thousand Pumps), a column furnace (1260 TCC) and a degasser (1260 Degassing). Separation was performed with a reverse phase Agilent Poroshell 120 model EC-C18 (100 mm x 3.0 mm, 2.7 µm) analytical column and set at 40 °C. HPLC-MS/MS analyzes were performed using gradient elution with eluent A (water + 5 mM ammonium formate + 0.1% formic acid: 85.0%) and eluent B (methanol + 5 mM ammonium formate + 0.1% formic acid: 15.0%). Analysis 0.400 mL/min flow rate, 400 bar pressure for 12 minutes (A: 50.0%, B: 50.0%), 30 minutes (A: 10.0%, B: 90.0%), 32 minutes ( A: 10.0%, B: 90.0%, and 35 minutes (A: 85.0%, B: 15.0%) [6].
1HNMR analysis
1HNMR spectra were obtained with an Agilent-600 MHz NMR spectrometer (Santa Clara, USA). CDCl3 or DMSO-d6 solvents were used in 1HNMR studies. 1HNMR spectra of WBTP extracts were detected using 64 data points with a spectral width of 9615 Hz and 64 pulse sequences and a frequency of 599.92 MHz, requiring approximately 1.8 s per sample [6].
Biological activity tests
Antioxidant activity
Total antioxidant activity was analyzed according to the method of Prieto et al [8]. This method is based on the reduction of Mo+6 to Mo+5 and the formation of the green phosphate/Mo (V) compound. In this method, 1 ml of reagent solution (28 mM sodium phosphate: 0.6 M sulfuric acid: 4 mM ammonium molybdate) was added to 0.1 ml of WBTP extract. The reaction medium was incubated in a sealed tube in a shaking water bath at 95 °C for 90 minutes. The mixture was then cooled to 25°C. The absorbance value was measured at 695 nm and the results were calculated by calculating the A0.5 (µg/mL) values [6].
The reducing power of WBTP extracts was determined according to the method of Oyaizu et al[9]. in this method, 50 µL of 0.2 M phosphate buffer (pH 6.6) and 50 µL of 1% K3Fe(CN)6 were mixed with 20 µL of WBTP extract. The mixture was left in a 50°C water bath for 20 minutes. then 50 µl of 10% trichloroacetic acid (TCA) was added to the mixture medium. 50 µL of the obtained supernatant was mixed with 10 µL of 0.1% FeCl3 and 50 µL of ddH2O. The absorbance value was measured at 700 nm and the results were determined by calculating the A0.5 (µg/mL) values [6].
Radical scavenging activity was analyzed at 517 nm by Blois and Noshad et al [10, 11]. In this method, 150 µL of extract and solutions of standard antioxidant substances were mixed with 50 µL of 0.1 mM DPPH˙ solution. then incubated for 60 minutes at room temperature and in the dark. Methanol (0.05 ml) was applied to prepare the blank sample in the same manner. The absorbance value was measured at 517 nm and the results were calculated as IC50 (ug/mL)[6].
ABTS+ radical scavenging activity, a method reported by Re et al and Noshad et al [11, 12]. implemented with a slight modification. 2.45 mM K2SO8 and 7 mM ABTS (1:2) were mixed and incubated in the dark at room temperature for 12-16 hours. After the mixture was diluted with ethanol to give an absorbance value of 0.700±0.020, 20 µL of different extracts or standard concentrations were mixed with 180 µL of ABTS•+ solution in a 96-well plate. The absorbance value was measured at 734 nm after 6 minutes and the result was determined as the IC50 value (µg/mL) [6].
Metal chelation activity was performed according to the method of Dinis and Madeira [13]. In this method, 20 µL of WBTP extract and 2.5 µL of 2 mM FeCl2 were thoroughly mixed. The reaction is initiated by adding 10 µL of 5 mM ferrosine. The final volume was made up to 200 µL with ethanol. The mixture was thoroughly mixed by vortexing and incubated at 25°C for 10 minutes. The absorbance value was measured at 562 nm and the results were determined by calculating the IC50 (µg/mL) values [6].
The superoxide anion radical scavenging activity was determined according to the Nishikimi and Yagi method[14]. The superoxide radical was obtained by the reduced nicotinamide adenine dinucleotide (NADH)-phenazine methosulphate (PMS) system by reduction of nitro blue tetrazolium chloride (NBT) and oxidation of NAD. In the experiment, NBT (156 µM, 62.5 µL), 62.5 µL of different concentrations of WBTP extract and antioxidant solutions and NADH (468 µM, 62.5 µL) were mixed thoroughly. The reaction was started by adding PMS (100 µM, 25 µL) to the reaction mixture, incubated and left at room temperature for 5 minutes. The absorbance value was measured at 560 nm and the results were calculated as IC50 (µg/mL) [6].
Enzyme inhibition capacity
The α-amylase inhibition of WBTP extracts was determined using the iodine-KI (potassium iodide) method by combining 82 µl of the extract or acarbose with 10 µl of 1 U of α-amylase (in 20 mM pH 6.9 potassium phosphate buffer) with good mixing. Samples were incubated at 37 °C for 10 minutes. The mixture was mixed thoroughly with 8 µl of 1% starch solution and the samples were again incubated at 37 °C for 12 minutes. The mixture was mixed homogeneously with 50 µl of 10% HCl and 15 µl of iodine-KI (2.5 mM iodine + 6.5 mM KI), and then the sample was kept in boiling water for 10 minutes. Absorbance values were measured at 620 nm and results were calculated as IC50 values (µg/mL) [6].
Lipase inhibition of the extracts was determined using the 4-nitrophenyl octanoate method [15]. in this method, 20 µL of extract or orlistat, 200 µL of 100 mM pH 8.2 Tris-HCl buffer, 20 µL of 1 mg/mL lipase (in 100 mM pH 8.2 Tris-HCl buffer) were mixed homogeneously. Instead of 20 µL of extract, 20 µL of extract solvent was used as a control. The mixture was homogeneously mixed with 20 µL of 5.1 mM p-nitrophenyl octanoate (in 100 mM pH 8.2 Tris-HCl buffer) and incubated at 37 °C for 30 minutes. The absorbance value was measured at 410 nm and the IC50 (ug/mL) values of the results were calculated[6].
The tyrosinase inhibition of the extract was determined using the L-DOPA (3,4-dihydroxy-L-phenylalanine) method [16]. İn this method, 10 µL of WBTP extracts or kojic acid, 20 µL of 200 µg/mL tyrosinase (in 0.1 M pH 6.8 potassium phosphate buffer) and 20 µL of 0.1 M pH 6.8 potassium phosphate buffer were mixed homogeneously and at 37 °C. It was incubated at C for 10 minutes. The mixture was then mixed so that 20 µL of 5 mM L-DOPA was homogeneous. Absorbance values were measured at 475 nm and the IC50 (µg/mL) values of the results were calculated [6].
α-glucosidase was obtained according to the method of Mayur et al. using p-NPG (p-nitrophenyl α-D-glucopyranoside) [17]. 10 µL of WBTP extract or acarbose, 25 µL of 0.2 U of α-glucosidase (in 20 mM pH 6.9 potassium phosphate buffer) were mixed homogeneously. The mixture was homogeneously mixed with 25 µL of PNPG and 20 µL of 20 mM pH 6.9 potassium phosphate buffer, then the Extracts were incubated at 37 °C for 12 minutes. Afterwards, the mixture was mixed homogeneously by adding 100 µL of 0.2 M Na2CO3. The absorbance was measured at 410 nm and the results IC50 (µg/mL) values were calculated [6].
The carbonic anhydrase was analyzed according to the method of Chanda et al [18]. Using the 4-nitrophenol acetate method, 60 µL of the extract or acetazolamide, 90 µL of 115 U carbonic anhydrase (in 0.05 M pH 7.4 Tris-SO4 buffer) were mixed homogeneously. The mixture was mixed so that 60 µL of 10 mM 4-nitrophenol acetate solution was homogeneous. The absorbance value was measured at 400 nm and the results IC50 (µg/mL) values were calculated [6].
The urease inhibition activity, 10 µL of WBTP extract or thiourea, 25 µL of 1 U of urease (in 100 mM pH 8.2 sodium potassium buffer) and 50 µL of 17 mM urea were mixed well, then the Extract was incubated at 30 °C for 15 minutes. 45 µL of phenolic reagent (0.1% (w/v) sodium nitroprusside and 8% (w/v) phenol) and 70 µL of basic reagent (4.7% (v/v) NaOCl and 2.5% (w/v) h) NaOH) ) was mixed homogeneously. Samples were incubated at 30 °C for 50 minutes. IC50 (µg/mL) values were calculated by measuring the absorbance at 630 nm [6].
Acetylcholinesterase, and butyrylcholinesterase were tested according to the method of Ozen et al [19]. İn this method, 150 µL of 100 mM Na-K buffer (pH 8.0), 10 µL of different concentrations (0.025-5.0 µg/mL) of WBTP extract solutions or galantamine, 20 µL of enzyme solution (0.03 U/mL, 100 mM pH 8.0, Na- K buffer) was mixed into the tubes sequentially. 50 µL of 3.3 mM DTNB and 10 µL of 1 mM acetylcholine iodide or butyrylcholine chloride were added to the Mixture, followed by incubation at 25 °C for 15 minutes. Absorption was measured at 412 nm and IC50 values were expressed as µg/mL [6].
Antibacterial activity
Antibacterial activity was compared with amoxicillin and tetracycline using the minimum inhibitory concentration (MIC) method [19]. Four different Gram-negative bacteria (Escherichia coli; ATCC 25,922, Pseudomonas aeruginosa; ATCC 15,442, Klebsiella pneumonia; ATCC 10,031, Salmonella enterica; ATCC 13,311) and four Gram-positive bacteria (Enterococcus faecalis; ATCC faecalis; ATCC 29,212, Bacphy aureus; ATCC 25,213 and Listeria monocytogenes; ATCC 35,152) were cultured on sterile Mueller-Hinton agar (MHA: 38 g/L ddH2O) medium in a sterile cabinet at 37 °C for 24 h, strain B. cereus at 30 °C 16 -18 hours incubated. Each microbial culture was homogenized with sterile saline (0.85% NaCl) solution and diluted to 0.5 McFarland turbidity standard (1.5×108 CFU/mL) [6].
The MIC method was tested using different concentrations of WBTP extracts in a cationic Muller-Hinton water (MHB) medium and expressed as µg/mL. Cationic MHB was prepared by adding MgCl2 (2 mg/mL) and CaCl2 (2 mg/mL). The sterile 96-well plate was diluted in the range of 16,384 µg/mL to 8 µg/mL for 100 µL of extracts or antibiotics. 1 mL of 0.5 McFarland bacterial solution was mixed with 9 mL of cationic MHB, and 5 µL of the solution was added to all wells. The plate was then incubated at +4 °C for two hours in the refrigerator. Incubation was only performed at 37 °C in less than 18 hours. only B. cereus was incubated at 30 °C for 16-18 hours. The agar plates were then examined with the naked eye for growth or turbidity and recorded [6].
DNA protection activity
DNA protective activity of WBTP extracts was determined by agarose gel electrophoresis [20-22]. In this method, a 1% agarose gel was prepared and 2 µL of ethidium bromide was added. The agarose gel was poured and allowed to solidify (at least 2 hours). The thickened gel was deposited into the electrophoresis tank. 1X TBE buffer was transferred to the electrophoresis tank.
Negative Control: Plasmid DNA (3 µL)+dH2O (6 µL)+glycerol (4 µL).
Positive Control 3: Plasmid DNA (3 µL)+dH2O (6 µL)+glycerol (4 µL)+H2O2 (1 µL)+UV (5 min).
for WBTP extracts: Plasmid DNA (3 µL)+WBTP extracts (5 µL)+glycerol (4 µL)+H2O2 (1 µL)+UV (5 min).
2 µl of loading dye was added to control and WBTP extracts. Control and WBTP extract (1000 µg/ml) were loaded into wells and run at 90 volts for 60 min. Imaging was done with a UV trans illuminator (320 nm, 8000 µW/ cm) [6].
In this context, the food supplement production and different cosmetics from extracts, especially for cosmetic products made using Anatolian plants and endemic natural plant sources, will be mentioned within the scope of the project for the purification of plant-based and valuable chemicals and their conversion into functional products. In addition, within the scope of this study, the production, extraction, purification and conversion processes into functional products will be presented together, using completely domestic plant resources, and the necessary components to promote domestic production will be emphasized.
The importance of production in cooperation with the University and Industry in the food supplements sector and the studies carried out in the context of transforming our country's biological resources into production will be explained.
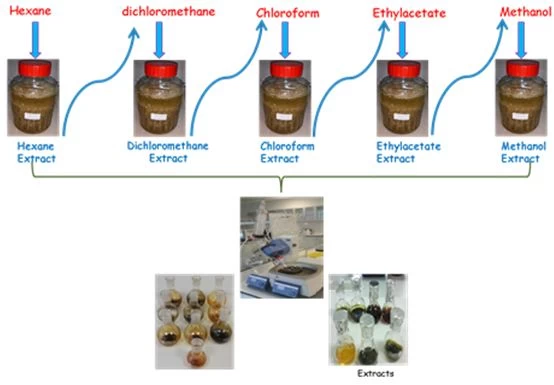
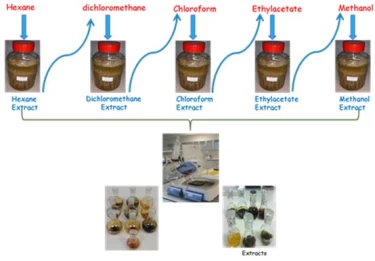
Figure 1 shows the extraction processes with increasing polarity from nonpolar to polar solvent, which is one of the extraction methods.1,2
In the lowest polarity such as hexane, lipids, steroids and hydrocarbons generally pass into the solvent, while highly polarized phenolic and flavonoid compounds pass into the solvent such as methanol.Since other solvents have different polarities, they allow molecules with different chemical structures, such as iridoids, to pass into the solvent.3
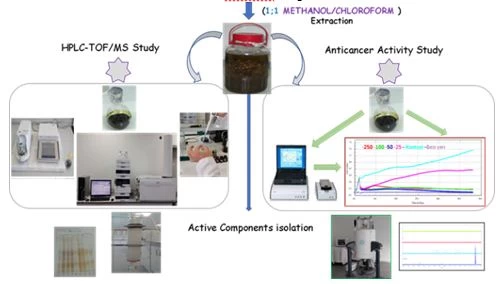
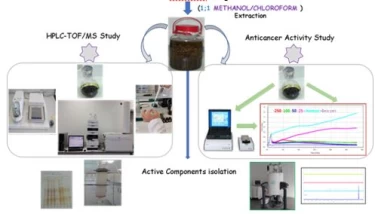
Figure 2. Solvent extraction of chloroform methanol mixture and then application of chromatographic separation methods.
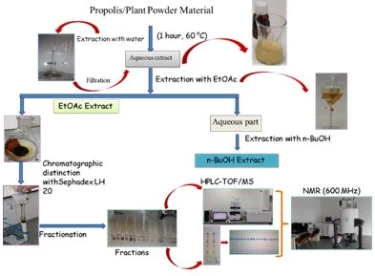
Figure 2 shows the extraction scheme with a mixture of methanol and chloroform. In such extractions, almost all of the secondary metabolites are taken into the solvent and it is aimed to purify the active components by chromatographic methods.Column chromatography with silica gel or Sephadexcolumns and TLC or preparative HPLC techniques are used for chromatographic separation methods.
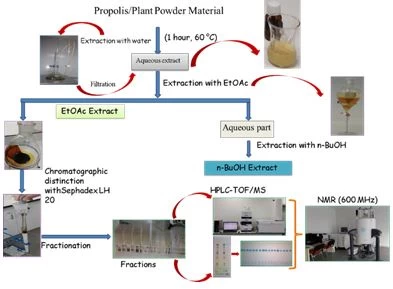
Figure 3 shows the extraction method with water. However, this method includes a very widely used method. In order for organic molecules to dissolve in water, heating is required. In this case, heat-resistant phenolic and flavonoid compounds can pass into the water. However, components that are not resistant to high temperatures,4 such as vitamin C, can break down.
Additionally, the thermal stability of vitamin C is within the temperature range of 30 to 90 °C. Melting point of ascorbic acid 190-192 Celsius.
In addition, many organic molecules are highly insoluble in water. For this reason, the maceration method with water has limited use and is not considered very suitable for industrial applications.
When the different extraction solvents were evaluated together, Ethyl alcohol, water and DMSO solvent (280.00, 268.17 and 284.84 mg GAE/g) were higher in total phenolic content, while DMSO-Water, propylene glycol and water (242.90, 248.28 and 238.60 mg GAE/g) were lower.
When the different durations were evaluated together, it was seen that water and dmso-water were effective in 15 days, while water-2 was more effective in 30 days. When all antioxidant tests were evaluated together, P5, P6, P7, P8, P9, P10 and P11 samples were found to be more effective in Eastern Anatolia, Mediterranean and Black Sea regions.
The solvents used in extractions are were effective for the extraction of organic acids such as caffeic acid, p-coumaric acid, t-ferulic acid, quercetin, apigenin, naringenin and campherolin large quantities.
Studies on transforming plant extracts into functional products and increasing their bioavailability as well as the purification and conversion of plant-derived and valuable chemicals into functional products are notenough. R&D studies should be focused on for the production of innovative, high value added and industrial scale plant extractions. It is essential to produce target-oriented and using new generation technologies. In order to make plant extracts usable, multi-functional and target-oriented production models should bedeveloped.In addition to R&D and P&D studies in plant extracts, new methods are required for industrial scaleproduction.
This work was supported by a grant from Cankiri Karatekin and Iğdır Universities.
1 V.C. Toreti et al., Evid Based Complement Alternat Med (2013) 697390
2 A. Salatino, M.L.F.Salatino, Apidologie 52(2) (2021) 312.
3 M. Surek et al., Journal of Ethnopharmacology 269 (2021) 113662.
4 L. Moreira et al., Food Chem Toxicol, 46(11) (2008) 3482.
5 S. Boulechfar et al., Z Naturforsch C J Biosci, 77(3-4) (2022) 105.
6 H.H. Omer, I. Demirtas, & T. Ozen, Journal of Food Measurement and Characterization (2022) 1.
7 E. Golmakani et al., The Journal of Supercritical Fluids 95 (2014) 318.
8 P. Prieto, M. Pineda, and M. Aguilar, Anal Biochem, 269(2) (1999) 337.
9 M. Oyaizu, The Japanese Journal of Nutrition and Dietetics 44(6) (1986) 307.
10 M.S. Blois, Nature 181(4617) (1958) 1199.
11 M. Noshad, and B. Alizadeh Behbahani, Food Science and Nutrition 9(3) (2021) 1625.
12 R. Re et al., Free Radic Biol Med. 26(9-10) (1999) 1231.
13 T.C. Dinis, V.M. Maderia, and L.M. Almeida, Arch Biochem Biophys. 315(1) (1994) 161.
14 M. Nishikimi, N. Appaji, and K. Yagi, Biochem Biophys Res Commun. 46(2) (1972) 849.
15 R. Trentin, et al., Algal Research 50 (2020) 101980.
16 N. Bibi Sadeer, K.I. Sinan, and Z. Cziáky, Molecules 27(6) (2022) 2000.
17 B. Mayur et al., L. J. Med. Plants Res. 4(15) (2010) 1547.
18 J. Chanda, and P.K. Mukherjee, Phytochem anal. 30(2) (2019) 148.
19 T. Özen, et al., Flavour and Fragrance Journal 35 (2020) 270.
20 A. Baiseitova, et al., Nat Prod Res. 35(5) (2021) 880.
21 A. Russo, et al., Phytother Res, 17(8) (2003) 870.
22 K. Sevgi, B. Tepe, and C. Sarikurkcu, Food Chem Toxicol 77 (2015) 12.
23 I. Demirtas I.H. Gecibesler, & A.S. Yaglioglu, Phytochemistry letters 6(2) (2013) 209.
24 I. Demirtaş et al., Food chemistry 136(1) (2013) 34.
25 F. Eser et al., Natural Product Research (2022) 1.
26 M.A. Demirci, et al., Food Chem. 269 (2018) 111-117.
İ. Demirtaş, New approaches in plant extraction methods, UNEC J. Eng. Appl. Sci. 2(2) (2022) 64-72
Anyone you share the following link with will be able to read this content:
This article is licensed under the Creative Commons Attribution ( CC BY 4.0 ) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
V.C. Toreti et al., Evid Based Complement Alternat Med (2013) 697390
A. Salatino, M.L.F.Salatino, Apidologie 52(2) (2021) 312.
M. Surek et al., Journal of Ethnopharmacology 269 (2021) 113662.
L. Moreira et al., Food Chem Toxicol, 46(11) (2008) 3482.
S. Boulechfar et al., Z Naturforsch C J Biosci, 77(3-4) (2022) 105.
H.H. Omer, I. Demirtas, & T. Ozen, Journal of Food Measurement and Characterization (2022) 1.
E. Golmakani et al., The Journal of Supercritical Fluids 95 (2014) 318.
P. Prieto, M. Pineda, and M. Aguilar, Anal Biochem, 269(2) (1999) 337.
M. Oyaizu, The Japanese Journal of Nutrition and Dietetics 44(6) (1986) 307.
M.S. Blois, Nature 181(4617) (1958) 1199.
M. Noshad, and B. Alizadeh Behbahani, Food Science and Nutrition 9(3) (2021) 1625.
R. Re et al., Free Radic Biol Med. 26(9-10) (1999) 1231.
T.C. Dinis, V.M. Maderia, and L.M. Almeida, Arch Biochem Biophys. 315(1) (1994) 161.
M. Nishikimi, N. Appaji, and K. Yagi, Biochem Biophys Res Commun. 46(2) (1972) 849.
R. Trentin, et al., Algal Research 50 (2020) 101980.
N. Bibi Sadeer, K.I. Sinan, and Z. Cziáky, Molecules 27(6) (2022) 2000.
B. Mayur et al., L. J. Med. Plants Res. 4(15) (2010) 1547.
J. Chanda, and P.K. Mukherjee, Phytochem anal. 30(2) (2019) 148.
T. Özen, et al., Flavour and Fragrance Journal 35 (2020) 270.
A. Baiseitova, et al., Nat Prod Res. 35(5) (2021) 880.
A. Russo, et al., Phytother Res, 17(8) (2003) 870.
K. Sevgi, B. Tepe, and C. Sarikurkcu, Food Chem Toxicol 77 (2015) 12.
I. Demirtas I.H. Gecibesler, & A.S. Yaglioglu, Phytochemistry letters 6(2) (2013) 209.
I. Demirtaş et al., Food chemistry 136(1) (2013) 34.
F. Eser et al., Natural Product Research (2022) 1.
M.A. Demirci, et al., Food Chem. 269 (2018) 111-117.