UNEC Journal of Engineering and Applied Sciences Volume 2 No 1, pages 26-32 (2022) Cite this article, ![]() 2067
2067
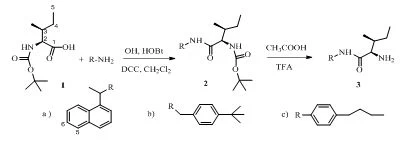
Due to the usage of synthetic compounds in many fields, the invention of a synthetic route and the synthesis of new compounds has attracted great interest [1-6]. Heterocyclic compounds have been employed in many areas. Nitrogen containing heterocycles have highly effective skeletons with a wide range of usages, remarkably in the synthesis of biologically active molecules [7-12]. Amides have attracted great importance in synthetic chemistry due to their versatility usage. Several methods for amides production have been developed for application in the pharmaceutical and medicinal industries. The treatment of carboxylic acid with amine is accepted as the effective method.
Density Functional Theory (DFT) with B3LYP / 6-31+G (d,p) basic set in the Gaussian 09w program presents the fully physical properties of molecules. Moreover, these values help interpreting the experimental spectra which provide the potential usage in drug development [13]. The aromatic aldehydes, significant, as well as intermediated compounds in synthetic chemistry were determined theoretically as physical and chemical properties that enable compounds to be used in various fields. The structural transformation of aromatic aldehydes such as 4-(N,N-dimethylamino)benzaldehyde [14], 3-quinolinealdehyde [15], was reported. Some corresponding compounds such as quinoline derivatives revealed the promising anti-cancer activity [16]. The diastereoselective syntheses of amides bearing two stereogenic centers in the 2,3 position with respect to nitrogen atom were achieved in a good yield [17].
Herein, the theoretical calculations of synthesized compounds were executed to determine drug potential.
Geometric optimization of molecules was performed in the Density Functional Theory (DFT) with B3LYP / 6-31+G (d,p) basic set in the Gaussian 09w program. The most stable and lowest energy conformer of the molecules is presented. The energies of the ground state geometry of the molecules and the dipole moment values, which are a measure of molecular polarity were calculated. Druglikeness features of synthesised compounds were calculated by Swiss ADME and Admet SAR predictor software [18, 19].
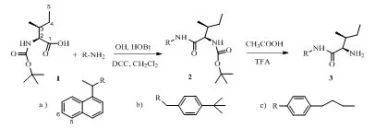
The synthesis method and spectral data were presented in the previous reported work [17].
Figure 1. Synthesis of bioactive compounds.
3.1. DFT calculation, geometric optimisation
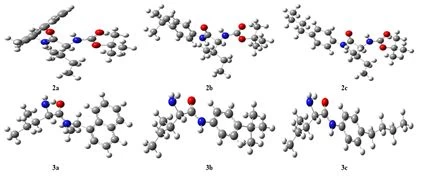
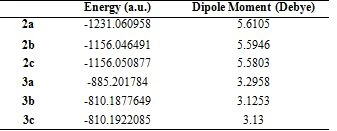
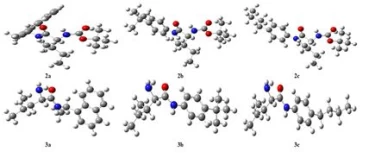
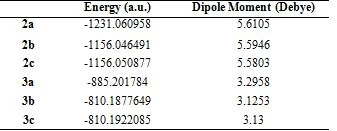
Geometric optimization of molecules was calculated in the Density Functional Theory (DFT) set B3LYP / 6-31+G (d,p). The most stable and lowest energy conformer of the molecules is given in Figure 2. The energies of the ground state geometries of the molecules and the dipole moment values, which are a measure of molecular polarity, are given in Table 1.
Figure 2. Optimized structure of synthesized molecules.
Table 1. The energy and dipole moment of compounds in DFT B3LYP / 6-31+G (d,p)
3.2. Molecular Orbital Properties
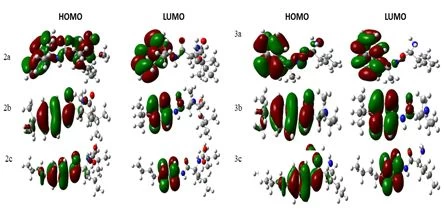
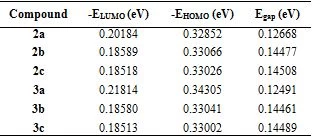
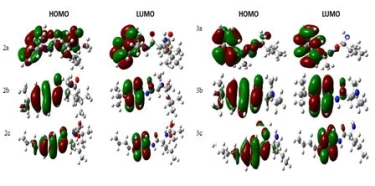
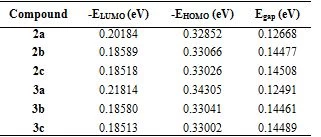
Geometric, electronic, and stable conformers of synthesised compounds were calculated using density functional theory (DFT) to prove the relationship between molecular structure and optoelectronic properties. All theoretical studies were carried out in the gas phase and the B3LYP/6-31G+(d,p) foundation set. The magnitude of the energy difference between HOMO-LUMO orbitals is considered a measure of the stability of the molecule (Figure 3). Stiffness is used as a measure to prevent intermolecular charge transfer. The molecule with high hardness has less charge transfer. Low chemical activity value and high kinetic stability indicate that the molecule is quite stable (Table 2).
Figure 3. Frontier molecular orbital (HOMO-LUMO) and related transition energy of compounds.
Table 2. Energies EHOMO, ELUMO and Egap for the studied molecules obtained by DFT/ B3LYP/6-31G+(d,p).
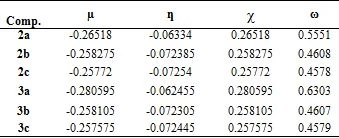
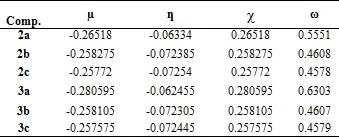
Chemical hardness (η) and softness (S) values of molecules can be calculated by energy differences of HOMO-LUMO orbitals obtained by DFT calculations (Table 3). The large HOMO-LUMO range gives high kinetic stability and low chemical reactivity; The low HOMO-LUMO range is important for low chemical stability, because it is convenient to remove electrons from the low energy HOMO orbital and add electrons to the high energy LUMO orbital [20]. Chemical reactivity indices such as electronegativity (χ), hardness (ƞ), electrophilicity index (x) and chemical potential (µ) were calculated by the following Eq (1):
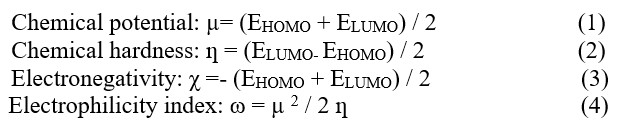
Table 3.Chemical reactivity indices, µ, ƞ, χ and ω for compounds obtained by B3LYP / 6-31G+ (d,p)
The HOMO-LUMO energy range of the synthesized compounds is low (Table 2). Based on the values given in Table 3, it is concluded that its chemical stability is low and it has high reactivity.
3.3. Molecular Electrostatic Potential Analysis
In order to determine the molecular electrostatic potential (MEP), electrostatically and nucleophilically active regions of the molecule, calculations were made in the gas phase in the B3LYP / 6-31G (d,p) basis set using the DFT method [21]. While the red color is suitable for electrophilic attack with high electron density, the blue region is suitable for nucleophilic attack with low electron density. Green areas are defined as neutral zones [22]. MEP displays molecular size, shape, and positive, negative, and neutral electrostatic potential regions simultaneously in terms of colour grading (Figure 4). It helps to interpret the biological recognition process and hydrogen bond interaction. It is very useful in studying molecular structure with physicochemical properties. The compound 6 have the highest electron density on the oxygen atoms and the positive charge density is high on the nitrogen atoms. There is electron density on the Naft groups in the compounds 2a and 3a and the negative charge density is delocalized to the aromatic structure. In the compounds 2b and 3b, it can be concluded that there is a negative charge distribution on the aromatic phenyl group.
Figure 4. Molecular electrostatic potential maps of all compounds
3.4. SwissADME Predictions
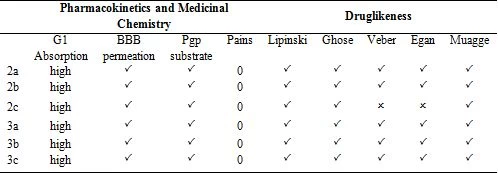
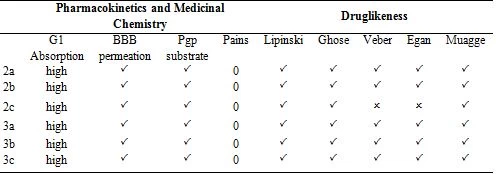
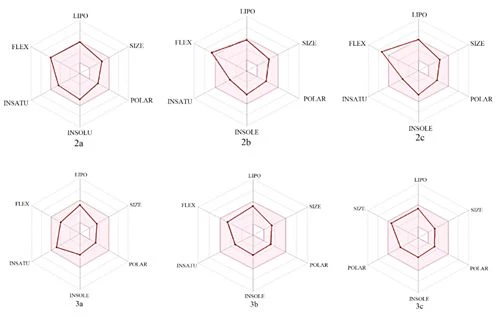
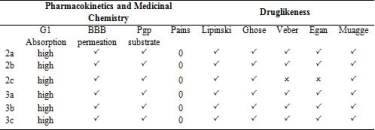
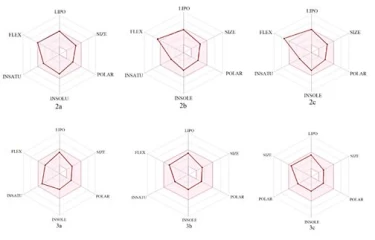
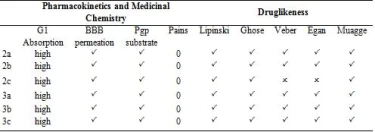
In today's technology, computational methods have been used to minimize the damage caused by chemicals to the environment. When designing a drug, some conditions are essential which are water solubility, water transport, intestinal lining, protein affinity, and toxicity. As a result of many years of studies on drugs, various drug similarity rules such as Lipinski rule, MDDR-like rule, Veber rule, Ghose filter, BBB rule, CMC-50-like rule, and Quantification of Drug Similarity (QED) have been developed. If the substances comply with these similarity rules, there is a high probability of drug likelihood (Table 4). Based on these possibilities, it has been observed that the compounds synthesized have drug efficacy. With the calculations made with the SwissAdme program, it is emerged that the compounds 2a, 2b, 3a, 3b, 3c obey the condition of Lipinski, Ghose, Veber, Egan, Muagge quarals, while the compound 2c does not comply with the Veber rule 10 <Rotors and Egar rule because XlogP3> 5. Synthesised molecules remained in the bioactivity radar, as seen in Table 4 and Figure 5 that show the drug properties of compounds.
Table 4. Druglikeness of the compounds
Figure 5. Bioactivity radars of drug candidate synthesized compounds.
3.5. ADMET Predictions
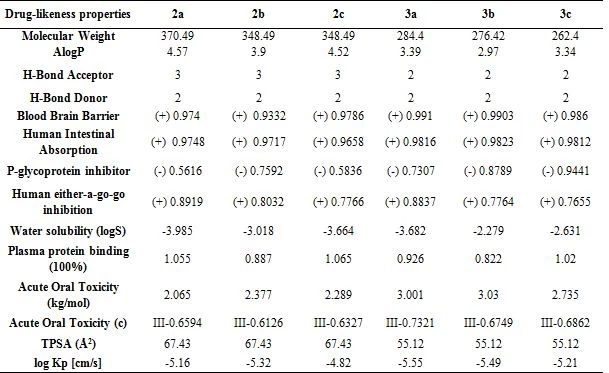
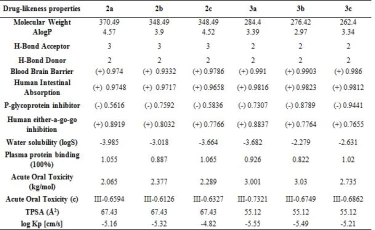
Lipophilicity and solubility are important parameters for the absorption of the drug by the body (Table 5). These pharmacokinetic parameters were obtained by the studies conducted by Lipinski et al [23]. According to the Lipinski rules; molecular weight should be equal to or less than 500 g/mol, hydrogen bond acceptor (HBA), total oxygen and nitrogen count should be less than or equal to 10, hydrogen bond donor (HBD), total OH and NH groups should be less than or equal to 5. The number of rotatable bonds (nRotb) should be equal to or less than 10a and the polarized surface area (TPSA) should be less than140 Ų. The synthesized compounds in the values obtained by drug similarity calculations were found to comply with the Lipinski rule.
Considering the Log S (ESOL: Estimating Aqueous Solubility) values obtained from the AdmetSAR calculation for the water solubility of the compounds, the newly synthesized molecules are dissolved in water. It was predicted by calculations that the synthesized compounds respond positively to the blood brain barrier (BBB) criteria and that the drugs can pass through the BBB. All drugs III. The category shows acute oral toxicity; this result helps us estimate its suitability for oral use as a drug. The AdmetSAR calculation helps us predict that the compounds are non-carcinogenic and suitable for tropic use. Not all drugs are P-glycoprotein inhibitors. P-glycoprotein inhibition can block the absorption, permeability, and retention of drugs (Table 5). However, all molecules exhibit non-inhibitory property for the human ether-a-go-related gene (hERG). HERG inhibition can lead to long QT syndrome so this aspect needs to be explored further [20].
Table 5. Selected pharmacokinetic parameters of compounds
*These parameters were determined with Swiss ADME and Admet SAR predictor.
The new carbamate derivatives (2a-c), pentanamide derivatives (3a-c) were synthesized with high yield. The desired products and intermediate products could be valuable for pharmaceutical industry. ADMET results suggest that all analogs are non-carcinogenic and safe for oral administration. In addition, it is suitable for use as a medicine because of its good water solubility, ease of passing through the intestinal membrane, and the ability to cross the brain barrier. Considering the rules used in theoretical studies in terms of drug likelihood, it is predicted that the drug likelihood of the compounds except compound 2c is high. In addition, when MEP maps are examined, it is predicted that the electron density is on oxygen atoms and is open to electrophilic attacks.
The numerical calculations reported in this article are performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA Resources)
1 I. Celik, I. Demirtas, M. Akkurt, R. Erenler, K. Guven, O. Cakmak, Crystal Research and Technology 38 (2003) 193.
2 I. Celik, M. Akkurt, I. Demirtas, R. Erenler, K. Guven, X-ray Structure Analysis Online 20 (2004) 89.
3 R. Erenler , O. Cakmak, Journal of Chemical Research (2004) 566.
4 O. Cakmak, R. Erenler, A. Tutar, N. Celik, Journal of Organic Chemistry 71 (2006) 1795.
5 R. Erenler, I. Demirtas, B. Buyukkidan, O. Cakmak, Journal of Chemical Research (2006) 753.
6 A. Tutar, R. Erenler, J.F. Biellmann, Journal of the Chemical Society of Pakistan 35 (2013) 1197.
7 R. Erenler, J.F. Biellmann, Tetrahedron Letters 46 (2005) 5683.
8 R. Erenler, J.F. Biellmann, Journal of the Chinese Chemical Society 54 (2007) 103.
9 R. Erenler, M. Uno, T.V. Goud, J.F. Biellmanna, Journal of Chemical Research (2009) 459.
10 A. Tutar , R. Erenler, Journal of the Chemical Society of Pakistan 34 (2012) 1526.
11 C.S. Unlu, A. Tutar, R. Erenler, Journal of the Chemical Society of Pakistan 34 (2012) 705.
12 J. Lu, I. Maezawa, S. Weerasekara, R. Erenler, T.D.T. Nguyen, J. Nguyen et al. Bioorganic and Medicinal Chemistry Letters 24 (2014) 3392.
13 L. Cluyts, A. Sharma, N. Kus, K. Schoone, R. Fausto, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 171 (2017) 207.
14 N. Kus, A. Sharma, R. Fausto, Tetrahedron 72 (2016) 5914.
15 N. Kus, M.S. Henriques, J.A. Paixao, L. Lapinski, R. Fausto, The Journal of Physical Chemistry A 118 (2014) 8708.
16 S. Okten, O. Cakmak, R. Erenler, O.Y. Sahin, S. Tekin, Turkish Journal of Chemistry 37 (2013) 896.
17 M. Sunkur, S. Aydin, T. Aral, B. Dag, R. Erenler, Organic Communications 14 (2021) 294.
18 T. Ince, R. Serttas, B. Demir, H. Atabey, N. Seferoglu, S. Erdogan, Journal of Molecular Structure 1217 (2020) 128400.
19 G. Kumar, R. Kumar, G. Ogruc-Ildiz, R. Fausto, A. Husain, Journal of Molecular Structure 1177 (2019) 33.
20 M.J. Hoque, A. Ahsan, M.B. Hossain, Biomedical Journal of Scientific & Technical Research 9 (2018) 7360.
21 P. Politzer, J.S. Murray, Reviews in computational chemistry (1991) 273.
22 S.M. Kawsar, M.A. Hossain, Turkish Computational and Theoretical Chemistry 4 (2020) 59.
23 C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Advanced drug delivery reviews 23 (1997) 3.
R. Erenler, B. Dag, B.B. Ozbek, Physical and bioactive properties of amide derivatives of L-isoleucine using theoretical calculation: drug-likeness, DFT and pharmacokinetics,UNEC J. Eng. Appl. Sci 2(1) (2022) 26-32
Anyone you share the following link with will be able to read this content:
This article is licensed under the Creative Commons Attribution ( CC BY 4.0 ) License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
I. Celik, I. Demirtas, M. Akkurt, R. Erenler, K. Guven, O. Cakmak, Crystal Research and Technology 38 (2003) 193.
I. Celik, M. Akkurt, I. Demirtas, R. Erenler, K. Guven, X-ray Structure Analysis Online 20 (2004) 89.
R. Erenler , O. Cakmak, Journal of Chemical Research (2004) 566.
O. Cakmak, R. Erenler, A. Tutar, N. Celik, Journal of Organic Chemistry 71 (2006) 1795.
R. Erenler, I. Demirtas, B. Buyukkidan, O. Cakmak, Journal of Chemical Research (2006) 753.
A. Tutar, R. Erenler, J.F. Biellmann, Journal of the Chemical Society of Pakistan 35 (2013) 1197.
R. Erenler, J.F. Biellmann, Tetrahedron Letters 46 (2005) 5683.
R. Erenler, J.F. Biellmann, Journal of the Chinese Chemical Society 54 (2007) 103.
R. Erenler, M. Uno, T.V. Goud, J.F. Biellmanna, Journal of Chemical Research (2009) 459.
A. Tutar , R. Erenler, Journal of the Chemical Society of Pakistan 34 (2012) 1526.
C.S. Unlu, A. Tutar, R. Erenler, Journal of the Chemical Society of Pakistan 34 (2012) 705.
J. Lu, I. Maezawa, S. Weerasekara, R. Erenler, T.D.T. Nguyen, J. Nguyen et al. Bioorganic and Medicinal Chemistry Letters 24 (2014) 3392.
L. Cluyts, A. Sharma, N. Kus, K. Schoone, R. Fausto, Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 171 (2017) 207.
N. Kus, A. Sharma, R. Fausto, Tetrahedron 72 (2016) 5914.
N. Kus, M.S. Henriques, J.A. Paixao, L. Lapinski, R. Fausto, The Journal of Physical Chemistry A 118 (2014) 8708.
S. Okten, O. Cakmak, R. Erenler, O.Y. Sahin, S. Tekin, Turkish Journal of Chemistry 37 (2013) 896.
M. Sunkur, S. Aydin, T. Aral, B. Dag, R. Erenler, Organic Communications 14 (2021) 294.
T. Ince, R. Serttas, B. Demir, H. Atabey, N. Seferoglu, S. Erdogan, Journal of Molecular Structure 1217 (2020) 128400.
G. Kumar, R. Kumar, G. Ogruc-Ildiz, R. Fausto, A. Husain, Journal of Molecular Structure 1177 (2019) 33.
M.J. Hoque, A. Ahsan, M.B. Hossain, Biomedical Journal of Scientific & Technical Research 9 (2018) 7360.
P. Politzer, J.S. Murray, Reviews in computational chemistry (1991) 273.
S.M. Kawsar, M.A. Hossain, Turkish Computational and Theoretical Chemistry 4 (2020) 59.
C.A. Lipinski, F. Lombardo, B.W. Dominy, P.J. Feeney, Advanced drug delivery reviews 23 (1997) 3.